Understanding Data Types and Basic Functions
Learning Objectives
- Describe what variables, vectors, and matrices are and how they can be manipulated in R.
- Use the built-in RStudio help interface to search for more information on R functions.
- Describe what a function is in R.
- Inspect the content of vectors in R and describe their content with class and str.
Recap Variables
A variable is a name that has a value associated with it
# Assigns a value to a variable
genome_size_mb <- 35
# Assigns a value to a variable and prints it out on the console
(genome_size_mb <- 35)
# Prints out the value of a variable on the console
genome_size_mb
Hint: tab key autocompletes.
Type geno and then tab. Let the computer do repetitious work. It’s easier and with fewer mistakes. Hint: A history of the commands you’ve run underHistoryin case you forgot to write something down
Functions
A function is a “canned script” that automates the processing of input and returns a value.
We will be using in-built function, however, other functions need to be first loaded (more on this later) before they can be executed .
For instance with sqrt: the input (the argument) must be a number, and the return value (in fact, the output) is the square root of that number. Executing a function or running it is called calling the function.
sqrt(49)
A function call is composed of two parts.
- Name of the function
- Arguments that the function requires to calculate the value it returns.
For instance as above:
sqrt() is the name of the function, and 49 is the argument.
We can also pass variables as the argument.
genome_length_mb <- 4.61
sqrt(genome_length_mb)
Functions can take multiple arguments. For instance, let’s round weight_lb to one decimal place.
Typing round() shows there are two arguments (pops up a yellow box).
If you know the function, but don’t know how to use it.
We can find out more details about the command of interest, we can use the help command too.
help(round)
Hint: Other helpful commands to explain the function are
?before the function e.g.?roundandargs(round)
However, if you are not sure what function is appropriate you can search all the help pages by running the command below… or simply searching google.
help.search("rounding of numbers")
There are two arguments: 1) the number to be rounded and 2) the number of digits
round(genome_length_mb, 1)
Functions return values, so as with other values and expressions, if we don’t save the output of a function, then there is no way to access it later.
Check the current value using the print command.
print(genome_length_mb)
To save the output of a function, we assign it to a variable.
genome_length_mb_rounded <- round(genome_length_mb, 1)
Exercise
- Now that R has genome_length_mb in memory, we can do arithmetic with it.
- For instance, we may want to convert this to the weight of the genome in picograms (for some reason).
978Mb = 1picogram.
- Divide the genome length and rounded genome length in Mb by 978.
Another function that we’ll use a lot is class(). All values and, therefore, all variables have types.
class(genome_length_mb)
Exercise
Check the data type of the following variables.
one <- 1.5
two <- “mega”
three <- FALSE
There are 6 data types:
characterfor string valuesnumericfor numberslogicalfor TRUE and FALSE (the boolean data type)integerfor integer numbers (e.g., 2L, the L indicates to R that it’s an integer)complexto represent complex numbers with real and imaginary parts (e.g., 1+4i)rawvalues store sequences of bytes, essentially representing raw binary data
Data Structures
We will be going through vectors, lists, matrixes, and dataframes.
Vectors
Let’s create a vector containing the model organisms.
model_org <- c("echerichia_coli", "homo_sapiens", "chlamydomonas_reinhardtii","drosophilia_melanogaster","schizosaccharomyces_pombe","Saccharomyces_cerevisiae","arabidopsis_thaliana","cavia_porcellus","xenopus_laevis","nothobranchius_furzeri","xenopus_laevis","rattus_norvegicus","danio_rerio")
In order to extract one or several values from a vector, we must provide one or several indices in square brackets, just as we do in math. R indexes start at 1.
So, to extract the 2nd element of model_org we type:
model_org[2]
We can extract multiple elements at a time by specifying multiple indices inside the square brackets as a vector. Notice how you can use : to make a vector of all integers and two numbers.
model_org[c(1,7)]
model_org[3:6]
model_org[10:1]
model_org[c(2, 8:10)]
Exercise
1) Select every other element in
model_org. Using model_org[c()]2) However, this is laborious. If we were dealing with a much longer vector- we can use the
seq()function to create sequences of numbers quickly. Use the help command to find how to form the elements in the vector you created above Fill in the blank to select the even elements of model_org using the seq() function.
List
Note that a vector is an “atomic” vector. An “atomic” vector has a homogeneous datatype in every element. A list is still a vector in R, but it’s not an atomic vector.
Three operators can be used to extract subsets of R objects.
[returns an object of the same class as the original.[[used to extract elements of a list or a data frame. It can only be used to extract a single element, and the returned object’s class will not necessarily be a list or data frame.Hint: If you apply
[to a list it always returns a list: it never gives you the contents of the list.- The $ operator is used to extract elements of a list or data frame by literal name. Its semantics are similar to that of [[.
(drosophilia <- list(model_org = TRUE, num_nobel_drosophilists = 9L, num_species = 3 * 500, nobel = c("Thomas Hunt Morgan","Hermann Joseph Muller")))
#> $model_org
#> [1] TRUE
#>
#> $num_nobel_drosophilists
#> [1] 9
#>
#> $num_species
#> [1] 1500
#>
#> $nobel
#> [1] "Thomas Hunt Morgan" "Hermann Joseph Muller"
As seen above, the advantage of a list is that it can:
- Be heterogeneous, i.e. can be of different datatype. They don’t need to be atomic vectors – you can stick a function in there!
- Have different lengths.
- Have names. Or not. Or some of both.
Exercise
How do we extract the name “Hermann Joseph Muller”? Choose one or more of the options below.
a.
drosophilia$nobel[2]b.
drosophilia[4][2]c.
drosophilia[[4]][2]d.
drosophilia[-1][1]
Matrixes
Matrixes are:
- two-dimensional organisation of an m*n array
- good for arithmetic operations
- only a single class of data
To form a simple matrix, we use the command below.
matrix(1:9, nrow = 3, ncol = 3)
However, when we want to fill the matrix row-wise, we can start using different arguments.
We will use the byrow argument to fill the matrix row-wise using boolean T/F. This is an example of a default argument, which means it will have an automatic value assigned, so it does not need to be specified, unlike a keyword argument.
x <- matrix(1:9, nrow=3, byrow=TRUE)
Exercise
Let’s check the dimensions using the
dimcommand orattributes. What is the difference between the outputs of both commands?attributes(x) dim(x)
To access elements of a matrix.
- Use square brackets
[indexing method - Elements can be accessed as
matrix[row_num,col_num]where row_num/column_num is a vector of the number assigned to the row/column of interest.
Select rows 2 and 3 and columns 1 and 3.
x[c(2,3),c(1,3)]
Exercise
Access:
1) the 2nd row of the matrix, x
2) the element from the 3rd row and 2nd column in the matrix, x
Extension:
Check the class of (1), (2), and (3) the matrix, x,
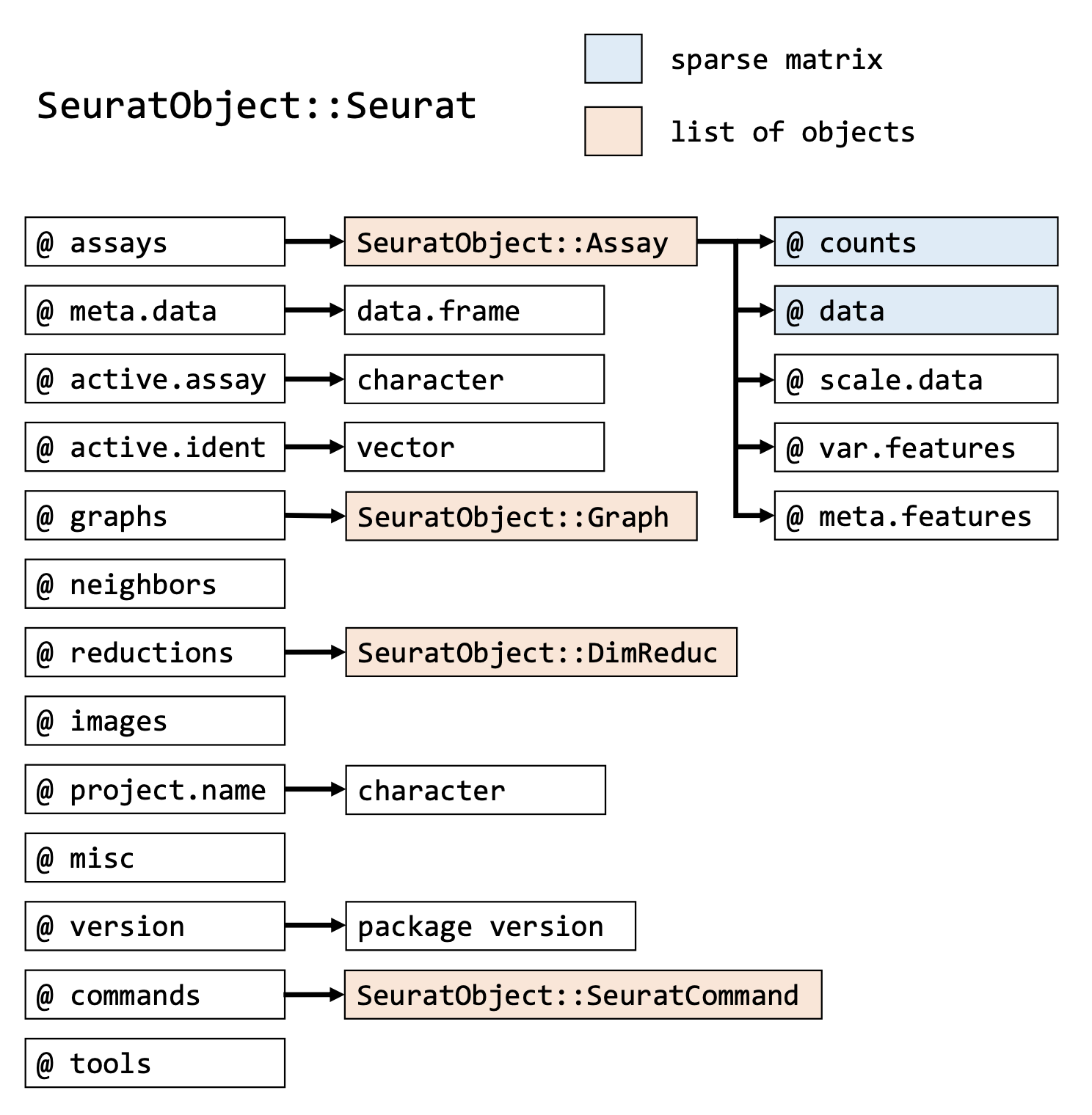
Note: For particular packages, there are custom R objects. These can combine all of the datatypes above to better streamline certain commands and pipelines. For example, Seurat has the format below.
Material adapted from (https://datacarpentry.org/R-genomics/01-intro-to-R.html) and (https://datacarpentry.org/semester-biology/materials/r-intro/)